







Les pathologies proches
Généralités
L’IRM a changé le diagnostic des Chiari ou des Syringomyélies et cela est un réel progrès. Cependant il est important que les dossiers soient étudiés par des neurochirurgiens spécialisés dans ces pathologies rares afin de bien déterminer la prise en charge. Ainsi des patients souffrent d’une Fente médullaire, n’auront pas de prise en charge Chirurgicale possible.
De même certaines pathologies sont associés à la Malformation de Chiari, soit qu’il est possible alors d’avoir une prise en charge adaptée comme dans les hydrocéphalies, soit que le Chiari est un symptôme d’une autre pathologie qui présente parfois d’autres symptômes similaires à ceux d’un Chiari évolué comme dans le Syndrome d’Edlers Danlos par exemple. On dit alors que le Chiari est un « acquis ». Là aussi, il est important de bien faire le différentiel de diagnostic.
LES FENTES MEDULLAIRES
L’examen d’un patient souffrant d’une syringomyélie nécessite des précautions particulières afin de diagnostiquer la pathologie pour assurer une prise en charge thérapeutique adaptée. Dans la grande majorité des cas, il est retrouvé une fine cavité (fente) pour laquelle le diagnostic différentiel doit faire la part entre les dilatations du canal centromédullaire et une véritable syringomyélie évolutive. Le canal centromédullaire est considéré comme une variante anatomique de la normale, présente à la naissance, qui se ferme progressivement et spontanément dans les 2 premières années de vie chez la grande majorité des sujets. Les fentes sont des fines cavités syringomyéliques qui peuvent présenter une évolution péjorative lentement progressive. Le diagnostic ne peut être confirmé qu’avec une surveillance clinique et radiologique régulière sur plusieurs années. La persistance du canal centromédullaire ne nécessite aucun traitement, s’agissant d’une simple variante anatomique.
C’est pourquoi il a été mis en place une étude (lire l’étude) pour déterminer le devenir des fentes intramédullaires.
De nombreux patients consultent pour des douleurs cervico-brachiales ou thoraciques, de type neuropathiques et rebelles aux traitements antalgiques habituels. Souvent la prise en charge chirurgicale n’est pas envisageable mais le parcours de prise en charge de la douleur est assez proche dans ce sens de celui de patients souffrant de syringomyélie.

L’HYDROCÉPHALIE
Le terme d’hydrocéphalie décrit une accumulation de LCR dans les ventricules du cerveau. Cette situation se produit habituellement suite au blocage des sentiers d’écoulement de LCR et peut avoir de nombreuses causes. Souvent, il est indiqué de traiter l’hydrocéphalie en premier lieu car la chirurgie au niveau de la jonction crânio-vertébrale peut aggraver l’hydrocéphalie. Le traitement de l’hydrocéphalie consiste habituellement à insérer un cathéter qui va détourner le LCR des ventricules cérébraux dans la cavité abdominale (shunt ou valve ventriculo-péritonéal(e)). D’autres techniques sont utilisées pour traiter l’hydrocéphalie comme l’intervention endoscopique (ventriculocisternostomie). Une équipe pluridisciplinaire, composée des spécialistes, neurologue, neurochirurgien, neuroradiologue et tout autre spécialiste impliqué dans le processus diagnostique et thérapeutique, devra prendre en considération toutes les options possibles et appropriées pour chaque cas.
La SYRINGOBULBIE
La syringomyélie est susceptible d’évoluer vers la syringobulbie, qui est une affection analogue, et correspondant à des complications au cours desquelles la cavité malformative se localise au niveau du bulbe, au lieu de la moelle épinière (comme dans la syringomyélie). Au cours de cette affection, qui ne comporte pas de lésions des organes eux-mêmes (pharynx, larynx, muscles du visage, oreille interne, oeil), ce sont les nerfs crâniens qui sont concernés, entraînant des troubles concernant les fonctions de ces nerfs à savoir :
- Perturbation de la déglutition (le fait d’avaler)
- Paralysie faciale
- Perturbation de la phonation (le fait d’émettre des sons)
- Anesthésie dans le territoire du nerf trijumeau (un des nerfs du visage)
- Syndrome labyrinthique (intéressant l’oreille interne), et accompagnant d’un nystagmus qui est un mouvement involontaire d’oscillation de faible amplitude et de rotation, du globe oculaire
L’évolution de cette affection est péjorative. En effet, il peut apparaître des perturbations de fonctionnement des centres nerveux situés dans le bulbe rachidien, et permettant normalement (physiologiquement) la régulation de la respiration, ce qui aboutit parfois à des syncopes, et au décès du patient.
L’ARCHNOÏDITE
L’arachnoïdite est un trouble causée par l’inflammation de l’arachnoïde, une des membranes qui entourent et protègent les nerfs de la moelle épinière.
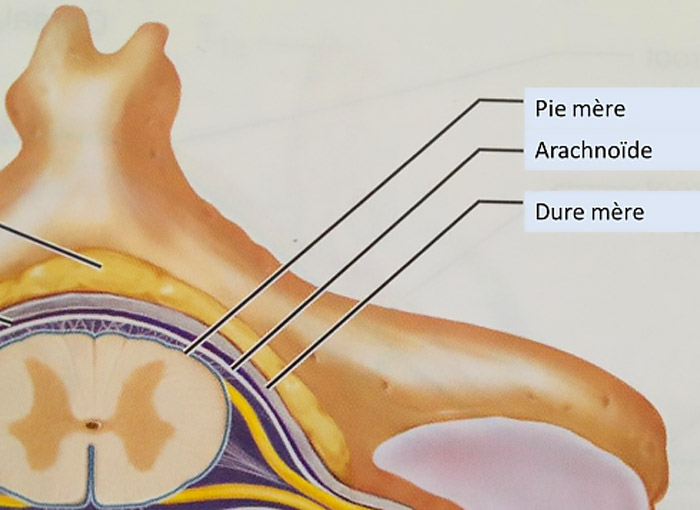
L’arachnoïde est l’une des trois méninges, située entre la dure-mère et la pie mère et séparée de la pie-mère par l’espace sous arachnoïdien (qui contient le liquide céphalorachidien).
Anatomie
L’arachnoïde est une membrane molle, ne contenant pas de vaisseaux. Les méninges sont constituées de trois enveloppes recouvrant le système nerveux central (cerveau et moelle épinière). Ce sont, de l’extérieur vers l’intérieur :
- La dure-mère ou pachyméninge (méninge dure) : méninge épaisse et fibreuse dont le rôle est de protéger l’encéphale (partie du système nerveux contenu dans la boîte crânienne : celui-ci comprend le cerveau, le cervelet et le tronc cérébral : segment supérieur de la moelle épinière. La dure-mère, située juste au-dessous de la boîte crânienne, sépare ces structures nerveuses de l’os
- L’arachnoïde est la membrane intermédiaire appelée ainsi pour sa ressemblance avec une toile d’araignée
- La pie-mère, très fine et vascularisée, qui recouvre directement le tissu nerveux
Lorsque la maladie progresse, les symptômes peuvent s’aggraver et devenir permanents.
Les causes de l’arachnoïdite
La cause de l’arachnoïdite est le plus souvent une infection généralisée à partir d’un point d’inoculation (foyer infectieux) pouvant être une sinusite ou une otite.
Dans certains cas l’arachnoïdite apparaît après un traumatisme, une hémorragie des méninges, une tumeur, l’introduction d’un corps étranger à l’intérieur des méninges (intervention) ou bien une méningite aiguë. La péridurale est parfois mise en cause.
Le cloisonnement peut aboutir à l’apparition d’une tension trop élevée du liquide céphalo-rachidien à l’intérieur de l’arachnoïde due à une insuffisance de circulation de ce liquide céphalo-rachidien ce qui aboutit quelquefois la formation de kystes.
Les symptômes
Les symptômes de l’arachnoïdite varient selon la localisation et l’étendue de la maladie :
- Maux de tête (céphalées)
- Épilepsie
- Atteinte des racines de la moelle épinière entraînant des troubles sensitifs
- Atteinte de la moelle épinière elle-même entraînant l’apparition de troubles moteurs à type de paraplégie ou de tétraplégie
- Mauvais fonctionnement de l’appareil génital et des sphincters entraînant une incontinence urinaire entre autres
L’arachnoïdite correspondant à l’inflammation de l’arachnoïde, aboutit quelquefois à la formation de kystes ou à un épaississement localisé entraînant la perte d’élasticité des tissus de certaines zones de l’arachnoïde.
Les traitements
Le traitement pour l’arachnoïdite est celui principalement celui de la cause (antibiotiques si la cause est infectieuse). Il n’existe aucun remède pour arachnoïdite installée. Les options de traitement pour les symptômes de l’arachnoïdite sont similaires à celles des autres états de douleur chronique. La plupart des traitements se concentrer sur l’amélioration de soulager la douleur et des symptômes qui nuisent à leurs activités quotidiennes. La chirurgie pour arachnoïdite est controversés, car les résultats peuvent être pauvres et ne fournissent qu’une aide à court terme.
LE SPINA BIFIDA
Le spina bifida ou dysraphisme spinal est une pathologie en lien avec une anomalie de développement du système nerveux, survenant au stade précoce de la grossesse. La fermeture du tube neural ne s’effectue pas de façon adaptée.On ne connait pas exactement la cause du spina bifida. La vitamine B9 prise en quantité suffisante en diminue le risque. La prise de certains médicaments en cours de grossesse peut l’augmenter.
Les patients souffrant de spina-bifida peuvent également être porteur de Malformation de Chiari 2 ou/et de syringomyélie
LES KYSTES DE TARLOV
Le kyste de Tarlov , également appelé kyste périneurale sacré ou péri-radiculaires est constitué de «poches» de liquide céphalo-rachidien (LCR) situées dans le canal rachidien, plus précisément entre le S4 et S1 au niveau des vertèbres. Ces structures ont d’abord été décrites par Isadore M. Tarlov, en 1938, au cours d’une étude de la borne de phylum ( filum terminale) lors de l’autopsie. Tarlov a noté de multiples kystes extradurales sur les nerfs dans la région du sacrum et / ou du coccyx. Jusqu’à présent, l’étiologie de cette maladie n’a pas été clairement élucidée. Cependant, il existe plusieurs hypothèses sur le facteur qui conduit à une augmentation du flux de LCR qui se présente dans les kystes, les obligeant à augmenter en taille, d’une forme asymptomatique ou symptomatique. Parmi ces causes sont des lésions qui se produisent dans le sacrum ou coccyx, résultant d’un accident d’automobile, ou d’un soulèvement d’objets lourds, la livraison et la région de l’analgésie péridurale.
Souvent, ces kystes présentent les mêmes symptômes que la syringomyélie et il n’a pas été décrit de correspondance entre les deux pathologies.
LE SYNDROME D’EHLERS DANLOS
Le syndrome d’Ehlers-Danlos est un groupe de maladies génétiques caractérisées par une anomalie du tissu conjonctif, c’est-à-dire des tissus de soutien.
Il existe différentes variantes de la maladie, la plupart présentent une hyperlaxité des articulations, une peau très élastique et des vaisseaux sanguins fragiles. Le syndrome n’affecte pas les capacités intellectuelles.
Le syndrome d’Ehlers-Danlos est une maladie génétique qui affecte la production de collagène, une protéine qui donne l’élasticité et la force aux tissus conjonctifs tels que la peau, les tendons, les ligaments, ainsi que les parois des organes et des vaisseaux sanguins. Des mutations dans différents gènes (par exemple ADAMTS2, COL1A1, COL1A2, COL3A1) seraient responsables des symptômes variables selon les différentes formes de la maladie.
Dans un certain nombre de cas (suffisamment pour que des équipes américaines travaillent sur le sujet) une Malformation de Chiari peut être associé. Une publication de l’ASAP (American Society Syringomyelia et Chiari Alliance Project a été traduite par l’UNSED (Union Nationale pour le Syndrome Edlers Danlos).

Le SYNDROME DE MARFAN
Le syndrome de Marfan est une maladie génétique protéiforme, qui peut rester silencieuse toute une vie avant d’être fatale si elle n’est pas traitée. En revanche, diagnostiquée et prise en charge assez tôt, elle n’empêche pas de mener une vie presque normale.
Exceptionnellement, une Malformation de Chiari est retrouvé chez de patients souffrant de syndrome de Marfan.
LA CRANIOSTENOSE
Une craniosténose est une fermeture prématurée d’une ou plusieurs sutures de la boite crânienne. On parle craniosténose non syndromique lorsque la craniosténose est isolée. Cependant, certaines craniosténoses peuvent être d’origine génétique. Dans certaines situations et afin d’assurer un bon suivi de l’enfant par la suite, une enquête génétique peut être nécessaire.
Un certain nombre d’enfants souffrant de craniosténose présentent une Malformation de Chiari associé, dite acquise. La prise en charge est souvent très différente de la Malformation Chiari isolée.
http://association-epitetes.fr/



